METHODOLOGY
|
Combining lineage-based and site-based sampling
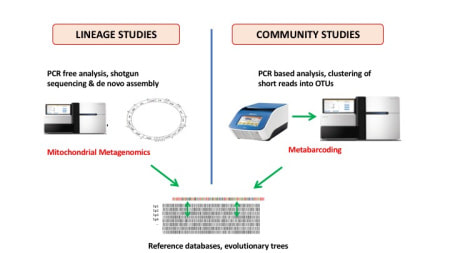
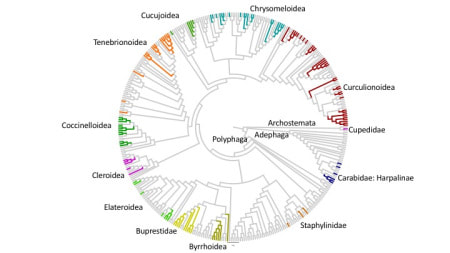
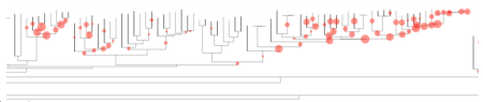
Thousands of specimens typically collected in passive arthropod traps can be processed for phylogenetic placement using full mitochondrial genomes assembled from shotgun sequencing, or can be studied with PCR-based ‘metabarcoding’. The latter is suitable for complex samples, to map short amplicons against the mitogenomes or for phylogenetic placement on the well supported tree from mitogenomes. Mitogenomes can be obtained by shotgun sequencing of total DNA. Short reads are assembled using standard genome assembly software, which preferentially generates contigs from high-copy number portions, such as mitochondrial DNA, in a process called 'genome skimming'. If applied to DNA mixtures from multiple specimens, mitochondrial genomes usually are assembled into separate contigs for each species in the mixture. Assembly may be less efficient if close relatives are present in the mixture, which also may lead to the formation of bioinformatically created chimeras. However, the process is sufficiently well developed to be useful in most routine applications. Alternatively, PCR can be used to obtain mitochondrial sequences, either generating multiple short amplicons for various genes or by long-range PCR. Assembled mitogenomes from local studies can be placed into the existing phylogenetic tree, e.g. obtained from Genbank data. See here for a sample of specimens from Borneo, placed on an existing tree of Coleoptera (Timmermans et al. GBE, 2016). With a tree in hand, short sequences from metabarcoding can be placed despite their low phylogenetic information. Various methods for phylogenetic placement are available. It is important to exploit the database in the most efficient way. For example, the RAPPAS (Rapid Alignment-free Phylogenetic Placement via Ancestral Sequences - https://github.com/blinard-BIOINFO/RAPPAS) technique places metabarcode sequences by first generating k-mers, which are placed on the branches or tips of a tree based on their matches to k-mers in the database whose position is determined by ancestral character reconstructions. In other words, the tree is represented as a set of k-mers of known phylogenetic position, against which the newly generated metabarcodes (new k-mers) can be placed. Once the database has been generated, the phylogenetic placement is extremely quick. A local sample can be placed relative to this existing phylogeny showing the probability of k-mer hits. |
|